Medical Device Registries
Advancing innovation and patient safety
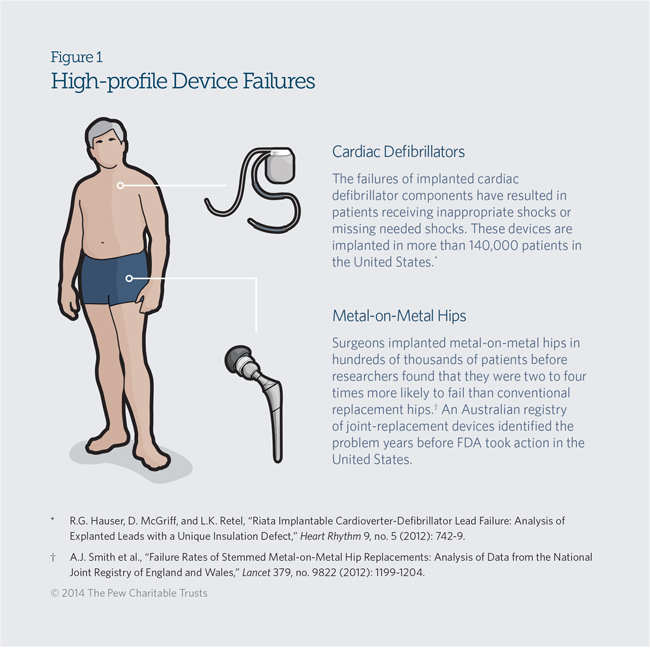
Recent, high-profile device failures underscore the need for better information on the post-approval performance of medical devices used in clinical care. (See Figure 1.) Medical device registries, which collect information on patients treated with specific products, are being used to solve this problem.
Download 'Medical Device Registries' (PDF)
Medical device registries
Manufacturers must present evidence that a device is safe and effective in order to gain FDA approval. Registries can capture more robust data beyond what FDA initially requires on a broader range of patients and in different clinical settings. In turn, clinicians can assess outcomes over time and more quickly detect problems with device safety. (See Table 1.)
Registries can also facilitate device innovation by collecting data more efficiently than traditional clinical studies, which can save manufacturers considerable time and money. For example:
- Data from a U.S. registry supported FDA’s decision to approve the use of an artificial heart valve in a larger population of patients than was originally authorized.1
- Scandinavian researchers used registries to enroll over 7,000 patients in a randomized clinical trial at less than a 10th the cost of a conventional randomized trial.2
Manufacturers and providers also may fulfill some of their regulatory obligations through registry participation. For example, manufacturers can use a registry to conduct FDA-mandated post-approval studies that collect information on a device’s performance once it enters clinical use. In addition, hospitals, which are required to report serious or life-threatening problems caused by medical devices, can use registries to document these issues for FDA.3

Next steps
A report from The Pew Charitable Trusts, the Medical Device Epidemiology Network, and the Blue Cross Blue Shield Association offers several recommendations to enhance the use of medical device registries in the United States.
FDA should collaborate with clinical societies, payors, providers, and manufacturers to determine which devices are most appropriate for a registry. These stakeholders should prioritize the use of registries based on whether there are important scientific questions about the device that a registry can efficiently answer, such as uncertainty about the long-term outcomes of implants. Device registries are particularly useful for monitoring patient outcomes when there is significant design variation in a class of products (e.g., hip implants are made from different materials and come in different sizes).
For registries to fulfill their potential, all stakeholders—especially clinicians, hospitals, FDA, and manufacturers—must work together to:
- Streamline registry data collection by limiting data fields, using standardized definitions, and integrating information from other sources—in particular, electronic health records and claims—to reduce the time and cost of reporting.
- Make information about registry governance, operation, and financing publicly available.
- Ensure that regulators, providers, patients, and manufacturers have access to registry findings in order to make evidence-based decisions.
- Disseminate registry findings to the public to facilitate informed decision-making.
- Gain clarity, particularly from federal agencies, on the interpretation of privacy and human subject protection laws for registries.
- Develop viable funding models to ensure the sustainability of registries.
Ultimately, expanding the use of registries will provide better information for patients and clinicians on the safety and effectiveness of devices and help innovative products to reach patients more quickly.
Endnotes
1 U.S. Food and Drug Administration, press release, “FDA Expands Access to Artificial Heart Valve for Inoperable Patients,” (Sept. 23, 2013), accessed Feb. 19, 2014, http://www.fda.gov/newsevents/newsroom/ pressannouncements/ucm369510.htm.
2 O. Frobert et al., “Thrombus Aspiration During St-Segment Elevation Myocardial Infarction,” New England Journal of Medicine, 369, no. 17 (2013): 1587; M.S. Lauer and R.B. D’Agostino, “The Randomized Registry Trial—The Next Disruptive Technology in Clinical Research?” New England Journal of Medicine 369, no. 17 (2013): 1580.
3 J.K. Kirklin and D.C. Naftel, letter from INTERMACS Registry to Institutional Review Board , March 28, 2014, accessed Aug. 22, 2014, http://www.uab.edu/medicine/intermacs/images/Opening page 4.0 info/Justification_Letter.pdf.



