Regenerative Medicine
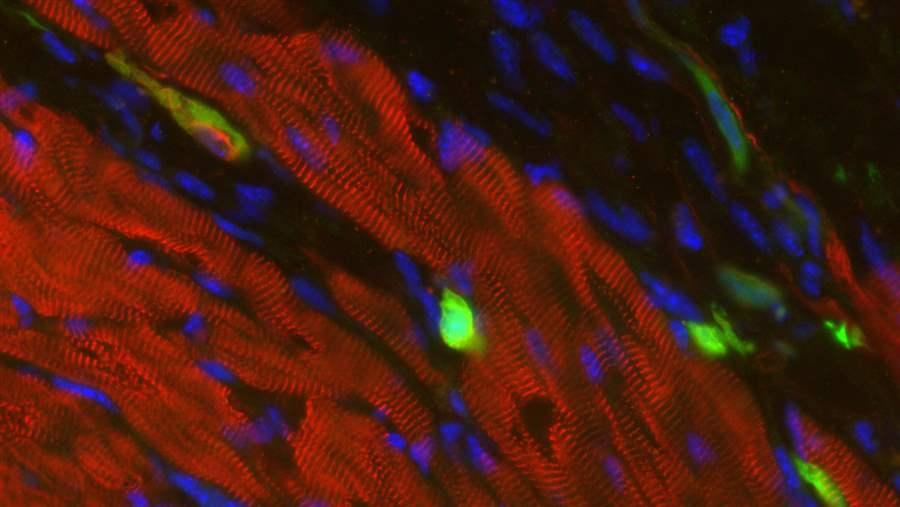
The cells in green are skin cells directly reprogrammed into heart muscle cells. The red indicates a protein that is unique to heart muscle. The technique used to reprogram the skin cells into heart cells could one day be used to mend heart muscle damaged by disease or heart attack.
California Institute of Regenerative Medicine
On May 5, 2017, this fact sheet was updated to clarify information regarding FDA regulation of regenerative medicine therapies.
Overview
Academic medical centers, pharmaceutical companies, and small private clinics are advancing therapies in regenerative medicine, a scientific field that is pioneering ways to regrow, repair, or replace human cells, tissues, and organs that have lost function due to damage or disease. The field holds promise for providing relief to patients suffering from organ failure, traumatic injuries, and chronic diseases.1 As these therapies are developed, policymakers will need to determine how to balance allowing access to these treatments while ensuring that they are safe and effective.
What treatments are being developed?
Researchers are developing treatments to regenerate damaged tissues in the body by stimulating previously irreparable tissues to heal themselves. For example:
- Tissue engineering has been used to create skin and bladder cells that, once transplanted, stimulate the growth of bone, connective tissues, and knee cartilage.
- Precursor cells, such as stem cells from adult tissues, are being studied for their potential to create engineered tissue.
- Researchers have developed a way to grow stem cells into mature bone cells that could potentially be transplanted into a patient.2
While some regenerative therapies have been approved by the Food and Drug Administration,3 others are administered in academic medical centers or private clinics. In recent years, clinics offering regenerative therapies (sometimes referred to as “stem cell clinics”) have proliferated in the United States. According to one study, at least 351 companies with 570 clinics marketed unapproved treatments in the U.S. in 2016 for conditions such as osteoarthritis, Alzheimer’s disease, autism, and injured spinal cords.4
How is regenerative medicine regulated?
Most drugs, devices, and biological products must be approved for safety and efficacy by FDA before they can be marketed. However, FDA exempts some human cells, tissues, and cellular and tissue-based products from the pre-approval requirement based on their risk.5
FDA divides these products into three regulatory categories:
- Requires premarket approval for safety and efficacy.
- Does not require premarket approval but is subject to provisions that help prevent the transmission of infectious disease.
- Does not require either of the above.
The last category applies only when cells, tissues, or products derived from them are removed and implanted into the same person during the same procedure, without any processing beyond rinsing, cleaning, or sizing. FDA has determined that the chances of a disease being transmitted during these procedures are no different from those typically associated with surgery,6 so there is no requirement to notify the agency. FDA has determined that this category applies in very limited circumstances.
However, clinics that engage in any “manufacturing” of cells, tissues, or products derived from them are subject to the agency’s regulation. FDA considers this to include “any or all steps in the recovery, processing, storage, labeling, packaging, or distribution of any human cell or tissue.”7 Manufacturers (or clinicians) creating these kinds of “lower” risk products (known as section 361 products8) need only register with FDA and comply with procedures intended to prevent the spread of infectious disease, provided they meet all the following criteria:
- The cells or tissues have been “minimally manipulated.” For “structural tissues” that physically support or serve as a barrier or cushion in the donor, such as tendons and ligaments, skin, or adipose (fat) tissue, processing cannot change the original characteristics of the tissue related to its utility to reconstruct or repair a damaged tissue. For cells or “nonstructural tissues,” such as nerves, glands, or lymph nodes, processing cannot alter the relevant biological characteristics of the cells or tissues.9
- The cells or tissues are intended for “homologous use.” The cells, tissues, or product must perform the same basic function in the recipient as in the donor. For example, a heart valve that is transplanted to replace a damaged heart valve would be considered homologous use because it performs the same basic function in the donor as in the recipient.10
- Manufacturing the products does not involving combining cells or tissues with other articles. With a few exceptions, including adding water or sterilizing agents, cells or tissues should not be combined with other materials that could raise new safety concerns.11
- The cells or tissues do not have a systemic effect throughout the body and do not depend on living cells for their primary function. Exceptions are those for “autologous use,” meaning cells or tissues transferred back into the same individual; those for “allogeneic use,” meaning cells or tissues from one individual transferred to another (in this case, it must be a first- or second-degree blood relative); and those for reproductive use.12
If a cell, tissue, or cellular and tissue-based product does not meet the criteria for a section 361 product or the same surgical procedure exception, it is treated as “higher” risk and must go through FDA premarket approval to demonstrate that it is effective and that its benefits outweigh its risks. Under a law passed in December 2016 as part of the 21st Century Cures Act, certain regenerative medicine therapies for serious or life-threatening conditions may be eligible for accelerated approval,13 which means that they may be reviewed on the basis of a surrogate measure of effectiveness, subject to post-approval trials.14 Sponsors of regenerative therapies with this priority designation may also be eligible for early and frequent meetings with FDA to discuss potential outcomes that would support the accelerated approval of their application.15
Technically, how a clinic or other provider of regenerative medicine therapies is regulated depends on what services that establishment offers. Some may be exempted entirely, some may be subject to the infectious disease controls in section 361, and others may be required to seek FDA approval. Practically, however, FDA does not have an accounting of who is offering these treatments, for what purposes, and under what controls, so even those clinics engaging in manufacturing practices may effectively be unregulated.
Implications
Regenerative medicine holds promise, but safety and effectiveness are likely to vary based on the therapy or product. FDA has approved treatments using cells derived from blood or bone marrow because safety and efficacy are well established. Increasingly, though, regenerative therapies derived from patients’ fat cells are being marketed based on limited evidence.16 Aside from treating blood and bone disorders, therapies for very few other health outcomes have been adequately tested for safety in clinical trials, and the large number of clinics already marketing these therapies warrants concern. In one instance, three patients became legally blind after undergoing an unapproved treatment for macular degeneration, in which the patients’ own cells were removed from fat and injected into their eyes.17 And very few regenerative medicine therapies have been adequately tested for effectiveness. Like other biologic products, FDA has developed pathways for accelerated approval of truly effective products intended to treat serious and life-threatening diseases. In order for patients and health care providers to understand the risks and benefits of regenerative medicine therapies, they should be evaluated based on adequate evidence.
Patients themselves may not understand which types of regenerative products and procedures are regulated by FDA, and the agency does not have a comprehensive list of clinics or manufacturers of these products or treatments. To protect the public, it will be important to have valid scientific evidence of their safety and effectiveness to avoid exposing patients to unknown risks and jeopardizing consumer confidence in this emerging field. Developing this evidence will be key to realizing the promise of regenerative medicine.
Endnotes
- Anthony Atala, “Regenerative Medicine Strategies,” Journal of Pediatric Surgery 47, no. 1 (2012): 17–28, doi:10.1016/j.jpedsurg.2011.10.013.
- National Institutes of Health, National Institute of Biomedical Imagining and Bioengineering, “Tissue Engineering and Regenerative Medicine,” accessed Nov. 1, 2016, https://www.nibib.nih.gov/science-education/science-topics/tissue-engineering-and-regenerative-medicine.
- FDA approvals do not designate products as regenerative, but products that may fall into that category include Carticel, which is a replacement for knee cartilage (source: Vericel Corp., “Carticel,” accessed March 20, 2017, http://www.carticel.com/); Provenge, which is a prostate cancer treatment (source: Valeant Pharmaceuticals International Inc., “Provenge,” accessed March 20, 2017, http://www.provenge.com/); Apligraf, which was approved to treat diabetic foot ulcers (source: Organogenesis Inc., “Apligraf,” accessed March 20, 2017, http://www.apligraf.com/); Gintuit, which is used for recovery from gum surgery (source: Organogenesis Inc., “Organogenesis Inc. Announces FDA Approval of Gintuit™ for Oral Soft Tissue Regeneration,” accessed March 20, 2017, http://www.organogenesis.com/news/press-release-announces-03122012.html); and Fibrocell, which replaces fibroblasts (source: Fibrocell Science Inc., “Fibrocell,” accessed March 20, 2017, http://fibrocell.com/).
- Leigh Turner and Paul Knoepfler, “Selling Stem Cells in the USDA: Assessing the Direct-to-Consumer Industry,” Cell Stem Cell 19, no. 2 (2016): 154–57, doi:10.1016/j.stem.2016.06.007.
- 21 CFR § 1271.
- U.S. Department of Health and Human Services, Food and Drug Administration, “Same Surgical Procedure Exception Under 21 CFR 1271.15(b): Questions and Answers Regarding the Scope of the Exception; Draft Guidance for Industry” (October 2014), http://www.fda.gov/downloads/BiologicsBloodVaccines/ GuidanceComplianceRegulatoryInformation/Guidances/Tissue/UCM419926.pdf.
- 21 CFR § 1271.3.
- Section 361 products are regulated solely under the authority of that provision of the Public Health and Service Act.
- U.S. Department of Health and Human Services, Food and Drug Administration, “Minimal Manipulation of Human Cells, Tissues, and Cellular and Tissue-Based Products; Draft Guidance for Industry and Food and Drug Administration Staff” (December 2014), http://www.fda.gov/downloads/BiologicsBloodVaccines/ GuidanceComplianceRegulatoryInformation/Guidances/ CellularandGeneTherapy/UCM427746.pdf.
- U.S. Department of Health and Human Services, Food and Drug Administration, “Homologous Use of Human Cells, Tissues, and Cellular and Tissue-Based Products; Draft Guidance for Industry and FDA Staff” (October 2015), http://www.fda.gov/downloads/BiologicsBloodVaccines/ GuidanceComplianceRegulatoryInformation/Guidances/Tissue/UCM469751.pdf.
- 21 CFR § 1271.10(a)
- Ibid.
- Pub. L. No. 114-225, § 3033 (a)(2).
- 21 CFR § 356 (c).
- Pub. L. No. 114-225, § 3033 (a)(4).
- Peter W. Marks et al., “Clarifying Stem-Cell Therapy’s Benefits and Risks,” New England Journal of Medicine 376, no. 11 (2017): 1007–9, doi:10.1056/NEJMp1613723.
- Ajay E. Kuriyan et al., “Vision Loss After Intravitreal Injection of Autologous ‘Stem Cells’ for AMD,” New England Journal of Medicine 376, no. 11 (2017): 1047–53, doi:10.1056/NEJMoa1609583.


ADDITIONAL RESOURCES

Article

Article







